Artificial intelligence (AI) is increasingly used in genomics to sift through vast amounts of genome data to identify potential therapeutic targets, despite the opaque nature of AI decision-making. To address this, Cold Spring Harbor Laboratory scientists have developed SQUID (Surrogate Quantitative Interpretability for Deepnets), a tool designed to enhance the interpretability of AI models in genomics.
SQUID, developed by scientists at Cold Spring Harbor Laboratory, improves the interpretability of AI in genomics by using a large library of DNA variants and the MAVE-NN program to analyze their effects.
This tool helps researchers make more accurate genetic predictions and supports hypothesis development for a better understanding of genomic functions.
SQUID Pries Open AI Black Box
Artificial intelligence continues to squirm its way into many aspects of our lives. But what about biology, the study of life itself? AI can sift through hundreds of thousands of genome data points to identify potential new therapeutic targets. While these genomic insights may appear helpful, scientists aren’t sure how today’s AI models come to their conclusions in the first place. Now, a new system named SQUID arrives on the scene armed to pry open AI’s black box of murky internal logic.
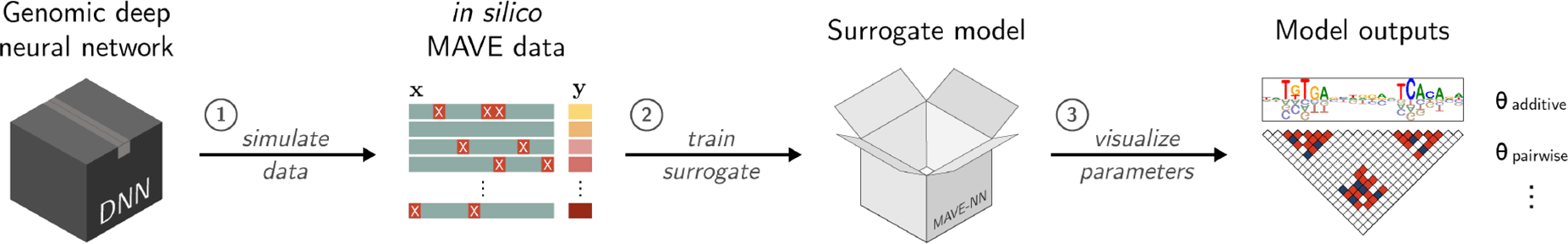
An illustration outlining the SQUID computational pipeline. Credit: Koo and Kinney Labs / Cold Spring Harbor Laboratory
SQUID: Enhancing AI Interpretability
SQUID, short for Surrogate Quantitative Interpretability for Deepnets, is a computational tool created by Cold Spring Harbor Laboratory (CSHL) scientists. It’s designed to help interpret how AI models analyze the genome. Compared with other analysis tools, SQUID is more consistent, reduces background noise, and can lead to more accurate predictions about the effects of genetic mutations.
How does it work so much better? The key, CSHL Assistant Professor Peter Koo says, lies in SQUID’s specialized training.
“The tools that people use to try to understand these models have been largely coming from other fields like computer vision or natural language processing. While they can be useful, they’re not optimal for genomics. What we did with SQUID was leverage decades of quantitative genetics knowledge to help us understand what these deep neural networks are learning,” explains Koo.

Evan E. Seitz, the lead author of this study, is a postdoc in the Kinney and Koo labs. Credit: Cold Spring Harbor Laboratory
SQUID works by first generating a library of over 100,000 variant DNA sequences. It then analyzes the library of mutations and their effects using a program called MAVE-NN (Multiplex Assays of Variant Effects Neural Network). This tool allows scientists to perform thousands of virtual experiments simultaneously. In effect, they can “fish out” the algorithms behind a given AI’s most accurate predictions. Their computational “catch” could set the stage for experiments that are more grounded in reality.
The Practical Impact of SQUID
“In silico [virtual] experiments are no replacement for actual laboratory experiments. Nevertheless, they can be very informative. They can help scientists form hypotheses for how a particular region of the genome works or how a mutation might have a clinically relevant effect,” explains CSHL Associate Professor Justin Kinney, a co-author of the study.
There are tons of AI models in the sea. More enter the waters each day. Koo, Kinney, and colleagues hope that SQUID will help scientists grab hold of those that best meet their specialized needs.
Though mapped, the human genome remains an incredibly challenging terrain. SQUID could help biologists navigate the field more effectively, bringing them closer to their findings’ true medical implications.
Reference: 21 June 2024, Nature Communications.
DOI: 10.1038/s42256-024-00851-5
Funding: Simons Foundation, National Institutes of Health, Alfred P. Sloan Foundation